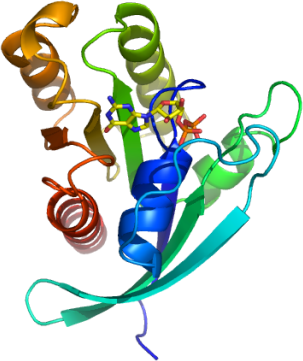
RRAS
PDB:2FN4
Revision
Revision Type:created
Revised by:created
Revision Date:created
Entry Clone Accession:RRASA-s001
Entry Clone Source:MGC
SGC Clone Accession:Tag:N-terminal hexahistidine tag that was removed by TEV cleavage
Host:BL-21(DE3)R3 phage resistant strain
Construct
Prelude:Sequence:smDPPPSETHKLVVVGGGGVGKSALTIQFIQSYFVSDYDPTIEDSYTKICSVDGIPARLDILDTAGQEEFGAMREQYMRAGHGFLLVFAINDRQSFNEVGKLFTQILRVKDRDDFPVVLVGNKADLESQRQVPRSEASAFGASHHVAYFEASAKLRLNVDEAFEQLVRAVRKYQEQELPPS
Residues sm are a result of the construct design.
Vector:pNIC28-BSA4
Growth
Medium:Antibiotics:Procedure:100 µL of BL21(DE3) competent bacteria were added to 96 wells of a sterile 96-well microtitre plate on ice. 3µL of each construct DNA was added to each well and mixed. The plate was left on ice for a further 30 minutes. The heat-shock procedure was done by transferring the plate to a 42°C water bath for 45 seconds and then returning it to sit on ice for 2 minutes. 100µL of LB medium was added to each well and the plate incubated at 37°C for 30 minutes. Each entire 203µL culture was plated out onto LB-Kan agar in a 5.5cm Petri dish. The plates were incubated at 37°C overnight. 12x 250mL flasks were prepared , each with 30mL of LB containing 50 µg/mL Kanamycin . Two colonies, from each transformation plate, were added to each starter culture. Flasks were left in a shaker at 200rpm and 37°C overnight. 19 mL of the starter culture was added to autoclaved 1litre TB medium that contained 50 µg/mL kanamycin. The flasks were placed in a shaker at 180rpm and 37°C. When the cell density reached an OD600 nm of approximately 1.0 the temperature was reduced to 20°C for 1 hr before induction with the addition of 1 mM IPTG. The cells were spun down (6500rpm, 15mins). The pellets were resuspended in 30 mL lysis/ binding buffer, transfered into 50 mL tubes and placed in a -80°C freezer.
Purification
ProcedureColumn 1: Low pressure IMAC
Buffers: Affinity wash buffer: 20 mM TRIS pH 8.0, 30 mM Imidazole, 200 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP; Affinity Elution Buffer: 20 mM TRIS pH 8.0, 250 mM Imidazole, 200 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP.
Procedure: Four Ni-Sepharose columns were prepared, 2 mL of 50 % Ni-Sepharose slurry was added to a clean, empty 15 mm diameter gravity column (providing a resin volume of 1 mL). The resin was equilibrated with 10 column volumes Lysis/Binding buffer. The lysates were allowed to drip through the column twice, and the flow through was collected .The columns were washed with 15 column volumes of Lysis/Binding Buffer (Wash1) and 25 column volumes of Wash Buffer (Wash 2). The bound protein wash eluted with 8 volumes of Elute Buffer (1 mL at the time) and 8 fractions were collected into eppendorf tubes and the samples stored overnight at
Enzymatic treatment: At this stage the purity of the protein was greater than 95 % based on SDS - PAGE analysis. The C-terminal hexahistidine tag was removed by TEV protease treatment. The TEV protease, a hexahistidine-tagged construct, was over-expressed and purified in-house to a final concentration of 2.5 mg/mL. Add 20 mL of the TEV protease was added to each fraction and left at 4°C overnight. The following steps were carried out to remove the cleaved products and TEV protease. Change buffer from Elution Buffer to 50 mM Tris pH 8, 150 mM NaCl using a 10-kD cutoff concentrator. Place 200 µL of 50 % Ni-NTA agarose in a 1.5 mL eppendorf tubes, add 1mL of 50 mM Tris pH 8, 150 mM NaCl mix, spin down and remove buffer. Repeat this resin wash step once. Add the TEV treated protein sample to the resin and mix for 30 min. Finally spin down resin and collect the supernatant which contains the cleaved RRASA.
Concentration: The concentration of RRASA was measured and 5 molar equivalents of GDP were added before concentrating to a 61 mg/mL. The conentrated protein was aliquoted into 50 µL volumes before freezing in the -80 o C freezer.
Extraction
ProcedureAfter thawing, the resuspended cells were lysed by passing through the Emusiflex C5 high pressure homogeniser. PEI (stock 5 %) was added to the homogenate to a final concentration of 0.15%. The cell debris, nuclei and DNA were spun down at 16,500 rpm for 45 min. The supernatant was collected.
Concentration:LigandMassSpec:Crystallization:Column 1: Low pressure IMAC
Buffers: Affinity wash buffer: 20 mM TRIS pH 8.0, 30 mM Imidazole, 200 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP; Affinity Elution Buffer: 20 mM TRIS pH 8.0, 250 mM Imidazole, 200 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP.
Procedure: Four Ni-Sepharose columns were prepared, 2 mL of 50 % Ni-Sepharose slurry was added to a clean, empty 15 mm diameter gravity column (providing a resin volume of 1 mL). The resin was equilibrated with 10 column volumes Lysis/Binding buffer. The lysates were allowed to drip through the column twice, and the flow through was collected .The columns were washed with 15 column volumes of Lysis/Binding Buffer (Wash1) and 25 column volumes of Wash Buffer (Wash 2). The bound protein wash eluted with 8 volumes of Elute Buffer (1 mL at the time) and 8 fractions were collected into eppendorf tubes and the samples stored overnight at
Enzymatic treatment: At this stage the purity of the protein was greater than 95 % based on SDS - PAGE analysis. The C-terminal hexahistidine tag was removed by TEV protease treatment. The TEV protease, a hexahistidine-tagged construct, was over-expressed and purified in-house to a final concentration of 2.5 mg/mL. Add 20 mL of the TEV protease was added to each fraction and left at 4°C overnight. The following steps were carried out to remove the cleaved products and TEV protease. Change buffer from Elution Buffer to 50 mM Tris pH 8, 150 mM NaCl using a 10-kD cutoff concentrator. Place 200 µL of 50 % Ni-NTA agarose in a 1.5 mL eppendorf tubes, add 1mL of 50 mM Tris pH 8, 150 mM NaCl mix, spin down and remove buffer. Repeat this resin wash step once. Add the TEV treated protein sample to the resin and mix for 30 min. Finally spin down resin and collect the supernatant which contains the cleaved RRASA.
Concentration: The concentration of RRASA was measured and 5 molar equivalents of GDP were added before concentrating to a 61 mg/mL. The conentrated protein was aliquoted into 50 µL volumes before freezing in the -80 o C freezer.
NMR Spectroscopy:
Data Collection:
Data Processing: