Casein kinase I (CKI) represents a unique group of serine/threonine protein kinases that is ubiquitously expressed in eukaryotic organisms. Seven mammalian CKI isoforms ( α, β, γ1, γ2, γ3, δ and ε ) and various splice variants have been identified to date. CKI family members contain a highly conserved N-terminal catalytic domain coupled to a variable C-terminal region that ranges in size from 40 to 180 amino acids. Casein kinases have been described as monomeric, constitutively active enzymes.
CSNK1G3 is widely expressed with high RNA levels detected in testis and levels found in brain, heart, liver, kidney and muscle . The characterization of the substrate specificity of CKI isoforms initially led to the identification of the canonical consensus sequence S/T(P)-X 1–2 -S/T, indicating that modification of serine or threonine residues by CKI requires the preceding phosphorylation of amino acid residues N-terminal of the target site. This requirement of a priming phosphorylation by another kinase restricted CKI to a function in the hierarchical phosphorylation of substrates. However, further studies revealed that a cluster of acidic amino acids N-terminal of the target serine/ threonine and an acidic amino acid in position n-3 could substitute for the phosphoamino acid efficiently. This non-canonical motif consisting of the sequence SLS has been shown to be recognized by CKI in combination with a cluster of acidic amino acid residues C-terminal of the phosphoacceptor site. The CKI substrates NF-AT and b -catenin exhibit such motifs, but phosphorylation of this non-canonical motif is 15–25 fold less efficient compared to the motif primed by a phosphoamino acid.
siRNAs targeting of CNK1G3 significantly enhances cell killing by A-443654 , an inhibitor fro the protein kinase Akt. Small molecules targeting CSNK1G3 in addition to Akt may thus exhibit increased efficacy and have the potential for improved therapeutic index. We have crystallized CSNK1G3 in complex with several inhibitors:
A cell-permeable 2,6,9-substituted purine (K00083; CGP74514A; N-(cis-2-Aminocyclohexyl)-N-(3-chlorophenyl)-9-ethyl-9H-purine-2,6-diamine) that acts as a potent, selective inhibitor of Cdk1/cyclin B (IC50 = 25nM). Reported to affect the activities of other kinases only at much higher concentrations (IC50 = 6.1µM, 125µM, and > 10µM for PKCa, PKA, and EGFR, respectively). This purine has been shown to induce mitochondrial damage and apoptosis (= 3µM) in several human leukemia cell lines and at lower concentrations (~ 1µM), an initial G2-M cell cycle arrest was observed in U937 cells, which eventually lead to apoptosis.
(SGC structure 2IZT)
Purvalanol A (K00227; 2-(1R-Isopropyl-2-hydroxyethylamino)-6-(3-chloroanilino)-9-isopropyl-purine) A potent, cell-permeable, and selective inhibitor of cyclin-dependent kinases (Cdks; IC50 = 4nM for Cdc2/cyclin B; IC50 = 70nM for Cdk2/cyclin A; IC50 = 35nM for Cdk2/cyclin E; and IC50 = 75nM for Cdk5/p35).
(SGC structure 2IZU)
A di-substituted diaminothiazole scaffold related to AG-12286 (K00577) which has been described as CDK4 inhibitors. (SGC structure 2IZS)
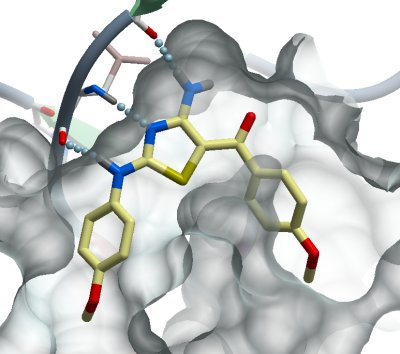
A related diaminothiazole with different substitution pattern at the two aryl rings (K00576) (this structure).
Structural comparison showed that both diaminothiazole inhibitors rearranged the glycine rich loop when compared to the purine type inhibitors. In addition to this structural rearrangement the extended region between the activation loop and the P1 loop rotated towards the solvent in structures in complex with the diaminothiazole type inhibitors.