Entry Clone Source: Site-directed mutagenesis |
Entry Clone Accession: n/a |
SGC Construct ID: ACVR1A-c096 |
GenBank GI number: gi|4501895 |
Vector: pFB-LIC-Bse. Details [ PDF ]; Sequence [ FASTA ] or [ GenBank ]
|
Amplified construct sequence:
TACTTCCAATCCATGCAAAGAACAGTGGCT
CGCGATATTACACTGTTGGAGTGTGTCGGG
AAAGGCAGGTATGGTGAGGTGTGGAGGGGC
AGCTGGCAAGGGGAAAATGTTGCCGTGAAG
ATCTTCTCCTCCCGTGATGAGAAGTCATGG
TTCAGGGAAACGGAATTGTACAACACTGTG
ATGCTGAGGCATGAAAATATCTTAGGTTTC
ATTGCTTCAGACATGACATCAAGACACTCC
AGTACCCAGCTGTGGTTAATTACACATTAT
CATGAAATGGGATCGTTGTACGACTATCTT
CAGCTTACTACTCTGGATACAGTTAGCTGC
CTTCGAATAGTGCTGTCCATAGCTAGTGGT
CTTGCACATTTGCACATAGAGATATTTGGG
ACCCAAGGGAAACCAGCCATTGCCCATCGA
GATTTAAAGAGCAAAAATATTCTGGTTAAG
AAGAATGGACAGTGTTGCATAGCAGATTTG
GGCCTGGCAGTCATGCATTCCCAGAGCACC
AATCAGCTTGATGTGGGGAACAATCCCCGT
GTGGGCACCAAGCGCTACATGGCCCCCGAA
GTTCTAGATGAAACCATCCAGGTGGATTGT
TTCGATTCTTATAAAAGGGTCGATATTTGG
GCCTTTGGACTTGTTTTGTGGGAAGTGGCC
AGGCGGATGGTGAGCAATGGTATAGTGGAG
GATTACAAGCCACCGTTCTACGATGTGGTT
CCCAATGACCCAAGTTTTGAAGATATGAGG
AAGGTAGTCTGTGTGGATCAACAAAGGCCA
AACATACCCAACAGATGGTTCTCAGACCCG
ACATTAACCTCTCTGGCCAAGCTAATGAAA
GAATGCTGGTATCAAAATCCATCCGCAAGA
CTCACAGCACTGCGTATCAAAAAGACTTTG
ACCAAAATTGATTGACAGTAAAGGTGGATA
|
Expressed protein sequence:
MGHHHHHHSSGVDLGTENLYFQ*SMQRTVA
RDITLLECVGKGRYGEVWRGSWQGENVAVK
IFSSRDEKSWFRETELYNTVMLRHENILGF
IASDMTSRHSSTQLWLITHYHEMGSLYDYL
QLTTLDTVSCLRIVLSIASGLAHLHIEIFG
TQGKPAIAHRDLKSKNILVKKNGQCCIADL
GLAVMHSQSTNQLDVGNNPRVGTKRYMAPE
VLDETIQVDCFDSYKRVDIWAFGLVLWEVA
RRMVSNGIVEDYKPPFYDVVPNDPSFEDMR
KVVCVDQQRPNIPNRWFSDPTLTSLAKLMK
ECWYQNPSARLTALRIKKTLTKID
Engineered Q207D mutation in bold and underlined. |
Tags and additions: MGHHHHHHSSGVDLGTENLYFQ*SM. cleavable N-terminal hexahistidine tag. |
Host: SF9 Spodoptera frugiperda Insect cells
|
Growth medium, induction protocol: Sf9 cells at a density of 2x106/ml were infected with recombinant ACVR1 baculovirus (virus stock P3; 1ml of virus stock/100 ml of cell culture). Cells were shaken at 110 rpm at 27°C in an Innova shaker. After 48 hours post-infection the cultures were harvested by centrifugation for 20min at 6000rpm. Cell pellets from each of three 1L cultures were resuspended in 15 ml binding buffer (50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 5 mM imidazole). Calbiochem protease inhibitor SET V was added to the cell suspension at a 1:2000 dilution and transferred to 50 ml tubes, and stored at -20°C. |
Extraction buffer, extraction method: The frozen cells were thawed and the volume increased to 200 ml with binding buffer. The cells were lysed using an Emulsiflex C5 homogeniser. The cell lysate was spun down by centrifugation at 21K rpm and 4°C for 1 h. The supernatant was recovered for purification. |
Column 1: Anion-exchange for Nucleic acid removal with DEAE cellulose (DE52, Whatmann)10 g of resin was suspended in 50 ml 1 M NaCl, and then applied onto a 2.5 x 20 cm column. The resin was then equilibrated with 50 ml binding buffer prior to loading the sample. |
Column 1 Buffers:
Binding buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 5 mM imidazole, 0.1mM TCEP
Wash buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 25 mM imidazole, 0.1mM TCEP |
Column 1 Procedure: The supernatant was first applied onto the column by gravity flow, which was followed by a wash with 50 ml wash buffer. The column flow-through and wash was directly applied onto a Ni-sepharose column. |
Column 2: Ni-Affinity Chromatography. 6 ml of 50 % Ni-sepharose slurry was applied onto a 1.5 x 10 cm column. The column was equilibrated with binding buffer (25ml). |
Column 2 Buffers:
Binding buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 5 mM imidazole, 0.1mM TCEP
Wash buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 25 mM imidazole, 0.1mM TCEP
Elution buffer: 50 mM HEPES, pH 7.5; 500 mM NaCl; 5% Glycerol; 50 to 250 mM imidazole, 0.1mM TCEP |
Column 2 Procedure: The flow-through from column 1 (DE52) was applied by gravity flow onto the Ni-sepharose column. The bound protein was eluted by applying a step gradient of imidazole - using 10 ml portions of elution buffer with increasing concentration of imidazole (50 mM, 100 mM, 150 mM, 250 mM). |
Enzymatic treatment: 0.1mg of TEV protease was added to the Ni-eluted protein to remove the tag. |
Column 3: Size Exclusion Chromatography - S200 HiLoad 16/60 Superdex run on ÄKTA-Express |
Column 3 Buffers: Gel Filtration buffer: 300 mM NaCl, 50 mM Hepes pH 7.5, 0.5mM TCEP |
Column 3 Procedure: Prior to applying the protein, the S200 16/60 column was washed and equilibrated with gel filtration buffer. The protein was concentrated to 4 ml using an Amicon Ultra-15 filter with a 10 kDa cut-off. The concentrated protein was directly applied onto the equilibrated S200 16/60 column, and run at a flow-rate of 1 ml/min. The protein was eluted at 90-108 ml. Fractions containing the protein were pooled together. |
Column 4: Ni-Affinity Chromatography. 0.75 ml of 50 % Ni-sepharose slurry was applied onto a 1.5 x 10 cm column. The column was equilibrated with binding buffer (15ml). |
Column 4 Buffers:
Binding buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 5 mM imidazole, 0.1mM TCEP
Wash buffer: 50 mM Hepes, pH 7.5; 500 mM NaCl; 5% Glycerol; 25 mM imidazole, 0.1mM TCEP
Elution buffer: 50 mM HEPES, pH 7.5; 500 mM NaCl; 5% Glycerol; 250 mM imidazole, 0.1mM TCEP |
Column 4 Procedure: The cleaved protein was passed through the column followed by 3ml binding buffer. It was then washed with 8ml wash buffer. Anything bound to the column was eluted with 15ml elution buffer. The pure ACVR1 protein at 2.6 mg/ml was flash frozen in liquid nitrogen and stored at -80°C. |
Column 5: Size Exclusion Chromatography - S200 HiLoad 16/60 Superdex run on ÄKTA-Express |
Column 5 Buffers: Gel Filtration buffer: 300 mM NaCl, 50 mM Hepes pH 7.5, 0.5mM TCEP |
Column 5 Procedure: Prior to applying the protein, the S200 16/60 column was washed and equilibrated with gel filtration buffer. 1ml of ACVR1 protein was thawed from -80°C storage and applied directly onto the equilibrated S200 16/60 column, and run at a flow-rate of 1 ml/min. The protein was eluted at 90-108ml. Fractions containing the protein were pooled together. |
Mass spectrometry characterization: The purified protein was homogeneous and had an experimental mass of 34494.5 (after TEV cleavage), closely matching the expected mass 34492.7. Mass was determined by LC-MS, using an Agilent LC/MSD TOF system with reversed-phase HPLC coupled to electrospray ionisation and an orthogonal time-of-flight mass analyser. Proteins were desalted prior to mass spectrometry by rapid elution off a C3 column with a gradient of 5-95% isopropanol in water with 0.1% formic acid. |
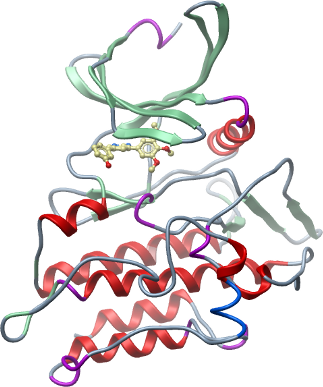
Crystallisation: Protein was buffered in 50 mM HEPES, pH 7.5, 300 mM NaCl, 10 mM DTT, 50mM L-arginine, 50 mM L-glutamate and concentrated to 7 mg/ml (calculated using an estimated extinction co-efficient of 58900). The inhibitor K02288a was added at 1 mM final concentration. Crystals were grown at 20°C in 150 nl sitting drops mixing 50 nl protein solution with 100 nl of a reservoir solution containing 1.6 M Na/KPO4, 0.1 M HEPES pH 7.5. On mounting crystals were cryoprotected with mother liquor plus 25% ethylene glycol and flash frozen in liquid nitrogen. |
Data Collection: Resolution: 2.15 Å resolution
X-ray source: Diamond Light Source, station I04, using monochromatic radiation at wavelength 0.9762 Å |